.svg)
Uni-FEP Workshop Successfully Held in Boston and San Francisco

Spotlight: Fully-automated Workflow in Uni-FEP
Free Energy Perturbation (FEP), a "gold standard" in global pharmaceutical research for its chemically accurate binding free energy predictions, faces challenges in traditional workflows. Over 80% of computational failures stem from improper preparation of initial protein-ligand complex structures, a time-consuming and labor-intensive process that poses challenges for users with limited expertise in CADD and structural biology.
- One-click construction of high-quality protein-ligand systems: Dr. Hang Zheng from Atombeat introduced the Uni-FEP Workflow on our Hermite platform , which fully automates the pipeline to build protein-ligand system that are ready for FEP calculation with only user-input protein structure and reference ligands. With various open-source and in-house tools, the workflow will perform protein structure preparation, ligand protonation state selection, and ligand structure alignment without any human intervention. Uni-FEP workflow yields high-quality starting structures for FEP calculation in a user-friendly and automated manner, dramatically lowering the barrier for users with different backgrounds.
- Comprehensive benchmarking builds user's confidence: Leveraging the automated Uni-FEP Workflow, researchers from Atombeat curated one of the largest benchmark datasets in this field, the Uni-FEP-Benchmark[1]. It comprises 376 targets and 6,341 ligand structures with experimental binding affinities, covering a wide range of protein families (e.g. kinase, protease and transferase) and real-world chemical challenges (e.g. scaffold-hopping, charge-changing). On this large-scale benchmark, the average RMSE between Uni-FEP predictions and experimental values was ~1.0 kcal/mol, demonstrating its robustness and accuracy. Prof. Olexandr Isayev and Prof. Junmei Wang acknowledged that this benchmark provided a rigorous validation for FEP techniques in real-world applications and would help to build user's confidence. Mr. Fillip Gusev further characterized it as the “best-practice guide” for researchers learning and mastering FEP.
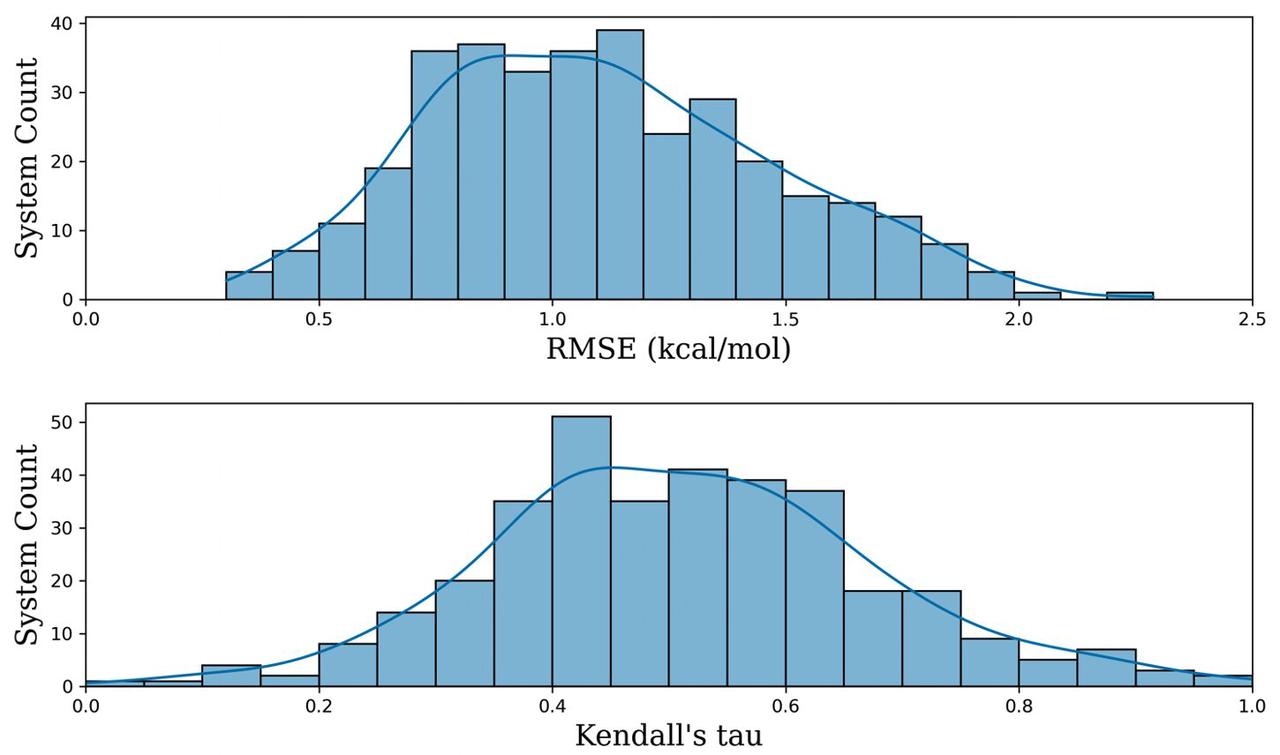
Figure 1. The distribution of RMSE and Kendall's tau of Uni-FEP predicted binding affinities and experimental values on systems in Uni-FEP-Benchmark datasets. Checkout our paper, Github-repo and Linkedin post for more information.
Key Improvements in FEP Accuracy: Sampling and Force Fields
The accuracy of FEP highly depends on the quality of sampling and force fields. Advances in these two topics bring Uni-FEP's accuracy on par with the state-of-the-art products.
- Sampling algorithm enhancements: Efficient sampling of slow degree of freedom in protein-ligand binding has always been a challenge in FEP-MD simulation. Dr. Xinyan Wang from Atombeat described advances in our Uni-FEP’s sampling strategies, including Terminal Flip Monte Carlo (TFMC) and Water Swap Monte Carlo (WSMC) methods. By performing a Monte Carlo move to flip terminal ligand functional groups during FEP-MD simulation, TFMC method can efficiently accelerate conformational sampling and evaluate contributions from different rotamer states on the fly, eliminating errors and additional cost introduced by existing methods like reweighting results from multiple simulations starting from different poses. WSMC addresses the issue of inadequate sampling of "buried waters" by frequently moving water molecules near and far away from the pocket through Monte Carlo attempts. With our in-house GPU implementation, the MC approaches can perform thousands of attempts with nearly no additional cost, thus boosting the sampling capabilities in Uni-FEP.

Figure 2. Illustration of two enhanced sampling techniques in Uni-FEP: Terminal Flip Monte Carlo (TFMC) for better side-chain rotamers sampling and Water Swap Monte Carlo (WSMC) for buried waters.
- Optimized Force Field for Ligands: Dr. Xinyan Wang also described our efforts of building more accurate force field (FF) for ligands. We designed a bespoke-fit workflow to optimize torsion parameters for any user-input ligand: the ligand will be first sliced to fragments, each of which is representative for a specific rotatable bond (torsion) and its parameters will be optimized against potential energy scan data with DPA2[2,3], an in-house foundation model to reproduce QM-level energy surfaces of biological molecules. The customized torsion parameters better describes binding conformation and yield accuracy improvements, especially demonstrated by a case-study system (PTP1B) where the simulation tended to sample an unrealistic unbound state with open-source GAFF2 force field.

Figure 3. Bespoke-fit workflow in Uni-FEP for torsion parameters with DPA-2, our in-house machine-learning potential.

Figure 4. (Left) Improved results with optimized force field in Uni-FEP, showcased by two systems. (Right) Refit torsion parameters fixed the unrealistic unbound stated sampled with GAFF2.
- Research Progress in Academia:
- Prof. Olexandr Isayev presented applications of the AIMNet2[3], a machine-learning potential model, across different aspects including potential energy surface fitting, conformational sampling, and binding affinity assessment. Dr. Xinyan Wang also introduced an on-going project about how Uni-FEP leverages AIMNet2 for force field fine-tuning and end-point free energy correction, yielding substantial improvements in FEP results.
- Prof. Bingqing Cheng emphasized the incorporation of physics in the development of machine-learning potentials. She highlighted the critical role of long-range electrostatic interactions in improving accuracy and transferability of existing ML potentials and demonstrated it with Latent Ewald Summation (LES), a model developed in her lab[4].
- Prof. Junmei Wang showcased his team’s high-precision atomic charge model for ABCG2[5] in reproducing physical properties and described IPSF[6], a rapid binding affinity prediction framework combining MM/GBSA with machine learning.
Panel Discussion: AI Agents + Computational Tools = The Future?
Participants discussed how to improve the usability and effectiveness of computational tools in drug design, with a particular focus on lowering usage barriers and integrating AI Agents.
- Lowering technical barriers: Dr. Ming Tommy Tang highlighted the necessity of reducing technical barriers in tool usage and parameter specification. He emphasized that it's critical to enable medicinal chemists to obtain reliable results without requiring software expertise, as tools must first be usable before they can deliver value.
- Flexible interface design: Professor Olexandr Isayev recommended that CADD tools should offer different type of interfaces for users with different backgrounds - graphical user interfaces (GUI) for medicinal chemists but programmatic and extensible APIs for CADD developers/experts to compose more complicated workflows.
- AI Agent integration potential: Dr. Emine Kucukbenli proposed connecting computational tools with large language models (LLMs) and AI Agents to reduce complexity in tool invocation and parameter tuning, enabling intelligent construction and optimization of drug discovery pipelines. Professor Junmei Wang also expressed strong anticipation for this direction.
- Forward-looking tool design: Dr. Yuxing Peng urged that new tools be architected from inception with built-in connectivity to active learning, reinforcement learning, and AI Agents, to fully unlock their potential.
AI Models: Crossing the “Usability” Threshold
Experts shared insights into elevating AI models from benchmarks to real-world applications in drug discovery.
- Surpassing the usability threshold: Dr. Zhiyong Sean Xie pointed out that, for AI models, marginal improvements on benchmarks are insufficient until they cross the "usability" threshold; only then will practitioners deploy them in real-world applications.
- Model reliability and physics constraints: Dr. Emine Kucukbenli emphasized the necessity of providing trustworthy uncertainty estimates so users can assess result reliability. She also proposed that integrating physical principles would be an effective way to address data scarcity.
- FEP and AI synergy: Mr. Fillip Gusev proposed combining FEP computations with AI modeling to efficiently build target-specific, reliable affinity predictors, thereby alleviating the speed bottlenecks of pure physical simulation.
- Simulation data empowering scientific AI: Dr. Yuxing Peng likened MD-generated data of biological systems to the datasets in autonomous driving development, arguing it is a valuable resource for training scientific AI models and should be fully leveraged.
Following the presentations, we conducted live demonstrations of Uni-FEP and received constructive, positive feedback from attendees. Our team also enjoyed all the thoughtful discussions with researchers from both industry and academia on frontier topics, ranging from physics-based modeling (force field development, enhanced sampling) to AI-driven methods. It was an honor for us, Atombeat Inc. to host the workshop, and we are grateful to all the speakers and participants for their time, insights, and engagement. We look forward to deepening these collaborations and advancing computational modeling tools together, with the shared goal of accelerating drug discovery as a collective effort.
References:
[1] Zou, Rongfeng, et.al., "Breaking Barriers in FEP Benchmarking: A Large-Scale Dataset Reflecting Real-World Drug Discovery Challenges." ChemRxiv. (2025): 2025-07
[2] Chang, Junhan, et al. "Efficient and Precise Force Field Optimization for Biomolecules Using DPA-2." arXiv preprint arXiv:2406.09817 (2024).
[3] Zhang, Duo, et al. "Pretraining of attention-based deep learning potential model for molecular simulation." npj Computational Materials 10.1 (2024): 94.
[4] Anstine, Dylan M., Roman Zubatyuk, and Olexandr Isayev. "AIMNet2: a neural network potential to meet your neutral, charged, organic, and elemental-organic needs." Chemical Science 16.23 (2025): 10228-10244.
[5] Kim, Dongjin, et al. "A universal augmentation framework for long-range electrostatics in machine learning interatomic potentials." arXiv preprint arXiv:2507.14302 (2025).
[6] He, Xibing, et al. "ABCG2: A Milestone Charge Model for Accurate Solvation Free Energy Calculation." Journal of Chemical Theory and Computation 21.6 (2025): 3032-3043.
[7] Zhai, Jingchen, et al. "In silico binding affinity prediction for metabotropic glutamate receptors using both endpoint free energy methods and a machine learning-based scoring function." Physical Chemistry Chemical Physics 24.30 (2022): 18291-18305.
Our mission is to democratize computational drug discovery tools and empower researchers and organizations worldwide.